```{r}
#| label: load-data
#| message: false
library(BiocFileCache)
bfc <- BiocFileCache()
raw.path <- bfcrpath(bfc, file.path(
"http://cf.10xgenomics.com/samples",
"cell-exp/2.1.0/pbmc4k/pbmc4k_raw_gene_bc_matrices.tar.gz"
))
untar(raw.path, exdir = file.path(tempdir(), "pbmc4k"))
library(DropletUtils)
library(Matrix)
fname <- file.path(tempdir(), "pbmc4k/raw_gene_bc_matrices/GRCh38")
sce.pbmc <- read10xCounts(fname, col.names = TRUE)
```Novedades RStudio. Quarto
2023-08-09
Novedades RStudio: Quarto
BIENVENIDOS
Presenta: Erick Cuevas Fernández




![]()

¿Por qué Quarto?
- Genera material interactivo utilizando Python, R, Julia y Observable.
- Difunde artículos, presentaciones, páginas web, blogs y libros de calidad profesional y reproducibles en formatos como HTML, PDF, MS Word, ePub, entre otros.
- Difunde saberes e innovaciones a lo largo de la organización mediante la publicación en Posit Connect, Confluence o otras plataformas de edición.
- Redacta usando el formato markdown de Pandoc, incorporando ecuaciones, menciones, enlaces internos, conjuntos de imágenes, destacados, estructuras complejas y otros elementos.
Vamos solo a trabajar directamente en RStudio
Se irán colocando aqui las instrucciones para que sirvan de guía.
HTML
- Abre un archivo en RStudio de Quarto.
- Elije de output HTML.
- Cambia la pre visualizacion al Viewer
Siguientes pasos
¿Qué notas de diferente con respecto a RMarkdown?
- Genera un Chunk para cargar los datos de PBMC4K.
Continua en tu archivo HTML
- Modifica el YAML para agregar en html: el flag code-link: true. ¿Qué es lo que sucede?
- Escribe texto para citar los datos utilizados de este link. ¿Cómo haríamos eso?
Siguiente ejercicio
- Escribe el siguiente chunk:
```{r}
#| label: gene-annotation
#| message: false
#| warning: false
library(scater)
rownames(sce.pbmc) <- uniquifyFeatureNames(
rowData(sce.pbmc)$ID, rowData(sce.pbmc)$Symbol
)
library(EnsDb.Hsapiens.v86)
location <- mapIds(EnsDb.Hsapiens.v86,
keys = rowData(sce.pbmc)$ID,
column = "SEQNAME", keytype = "GENEID"
)
set.seed(100)
e.out <- emptyDrops(counts(sce.pbmc))
sce.pbmc <- sce.pbmc[, which(e.out$FDR <= 0.001)]
```Nuestra primera tabla
- Escribe en tu HTML: “Table 1 es una tabla que nos muestra los outliers de las primeras 20 células”
¿Cómo hacemos el hipervinculo de Table 1?
Nuestra primera tabla
| Outliers | ||||
| Outliers de las primeras 20 células | ||||
| Total | LogProb | PValue | Limited | FDR |
|---|---|---|---|---|
| 1 | NA | NA | NA | NA |
| 0 | NA | NA | NA | NA |
| 1 | NA | NA | NA | NA |
| 0 | NA | NA | NA | NA |
| 1 | NA | NA | NA | NA |
| 1 | NA | NA | NA | NA |
| 0 | NA | NA | NA | NA |
| 0 | NA | NA | NA | NA |
| 0 | NA | NA | NA | NA |
| 1 | NA | NA | NA | NA |
Nuestro primer plot
Agrega el chunk y la lista a tu HTML y observa lo que sucede.
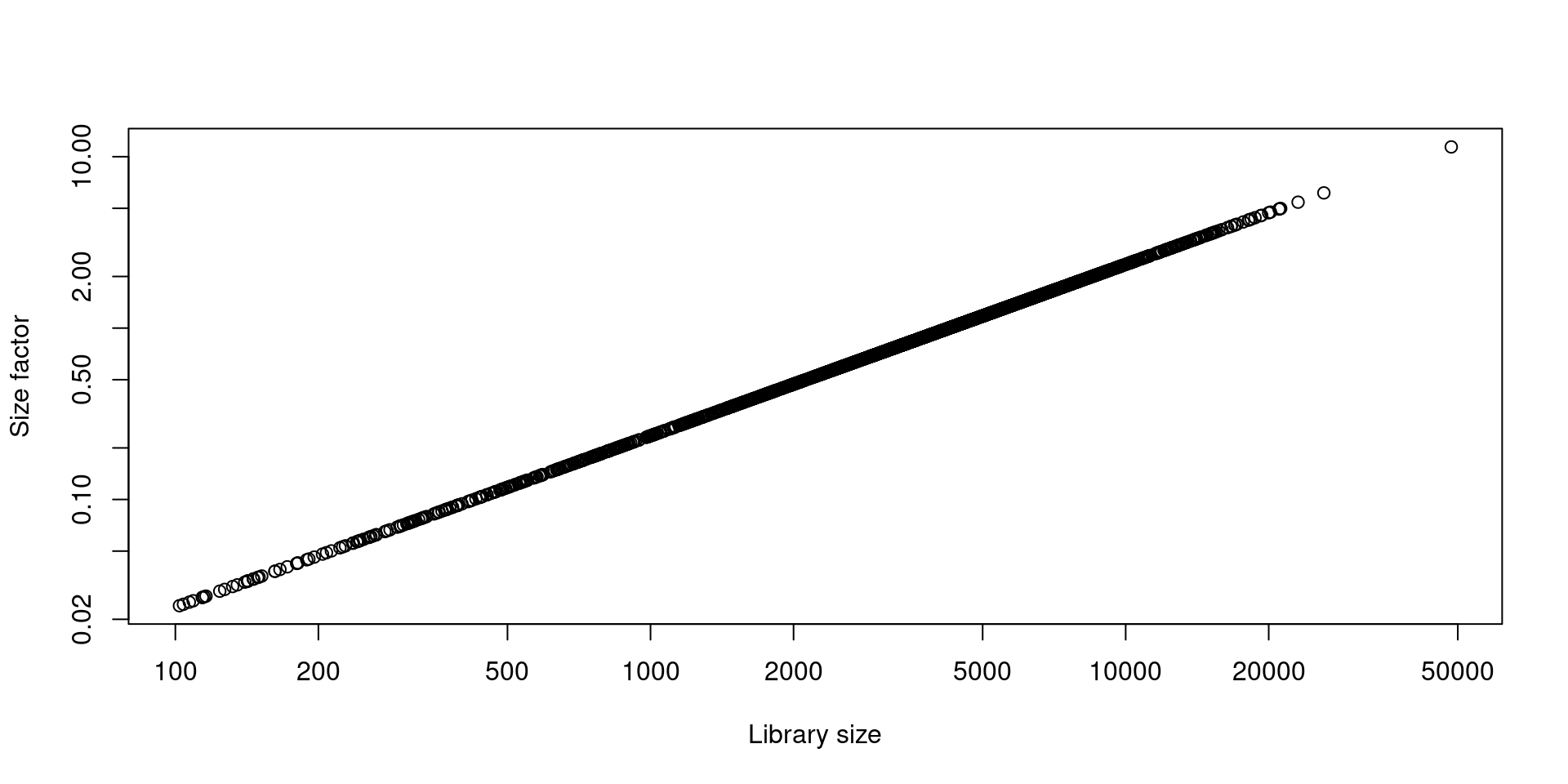
```{r}
#| label: fig-p1
#| fig-cap: Tamaño de libreria
#| output-location: slide
#| message: false
## Calculando el tamaño de las librerias
lib.sf.pbmc <- librarySizeFactors(sce.pbmc) # <1>
ls.pbmc <- colSums(counts(sce.pbmc)) # <2>
plot(ls.pbmc, lib.sf.pbmc, log="xy", # <3>
xlab="Library size", ylab="Size factor") # <3>
```- Estimar factores de normalización
- Calcular el tamaño de las librerias
- Plot
Nuestro primer plot
Vamos a cambiar a presentacion
- Cambia el YAML del output a revealsjs
- Agrega chalkboard: true
¿Qué sucedió?
Artículos
Para esta sección vamos a ir a quarto-journals.
El fin
MUCHAS GRACIAS

EBM 2023